Dear CADET team, I want to estimate the NU, SIGAMA and KA of the main component charge through the experimental data of different elution gradients, however, I have met some problems in this process, and I hope you can answer them
Here’s how I did it:
Since I do not know the initial values of these data, I extract some data from the literature, for example, the value of NU is set between 5-20, the value of SIGAMA is set between 20-60, the initial value of KA=1, NU=20, SIGAMA=40, the score of Shape is selected, and the search strategy of UNSGA3 with MCMC is used.
First of all, I set a large search range and got the initial search result, which was quite different from my initial value and because of the high characteristic charge value, there was a large sample peak in the elution curve, which was not reflected in my experimental data.
parameter1 = Dict()
parameter1.transform = ‘auto’
parameter1.component = 1
parameter1.bound = 0
parameter1.location = ‘/input/model/unit_001/adsorption/SMA_NU’
parameter1.min = 1
parameter1.max = 100
parameter2 = Dict()
parameter2.transform = ‘auto’
parameter2.component = 1
parameter2.bound = 0
parameter2.location = ‘/input/model/unit_001/adsorption/SMA_SIGMA’
parameter2.min = 1
parameter2.max = 1000
parameter3 = Dict()
parameter3.transform = ‘auto’
parameter3.component = 1
parameter3.bound = 0
parameter3.location = ‘/input/model/unit_001/adsorption/SMA_KA’
parameter3.min = 1e-5
parameter3.max = 1e5



Therefore, I took the search result of this time as the initial value, set the search scope as the literature reference value, and kept the other Settings unchanged with the first time. The fitting results are as follows,
parameter1 = Dict()
parameter1.transform = ‘auto’
parameter1.component = 1
parameter1.bound = 0
parameter1.location = ‘/input/model/unit_001/adsorption/SMA_NU’
parameter1.min = 5
parameter1.max = 20
parameter2 = Dict()
parameter2.transform = ‘auto’
parameter2.component = 1
parameter2.bound = 0
parameter2.location = ‘/input/model/unit_001/adsorption/SMA_SIGMA’
parameter2.min = 20
parameter2.max = 60
parameter3 = Dict()
parameter3.transform = ‘auto’
parameter3.component = 1
parameter3.bound = 0
parameter3.location = ‘/input/model/unit_001/adsorption/SMA_KA’
parameter3.min = 1e-5
parameter3.max = 1e5

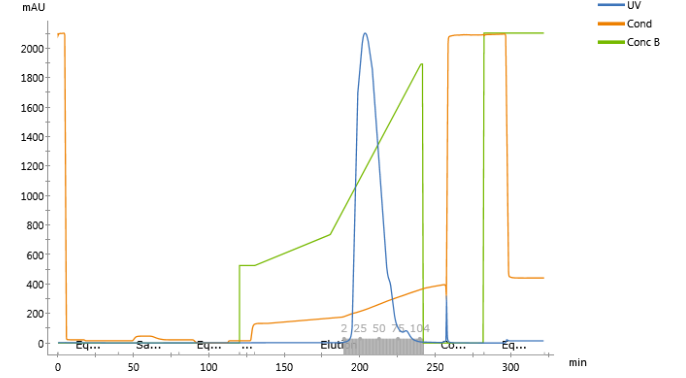
As it can be seen from the corner picture of the folder, is it because I have too large a range of SIGMA values that lead to multiple peaks? Now that you can see that the shape of my experimental data is very similar to that of the simulated data, how should I set the range and optimize the parameters next since I’ve tried a number of methods and I can’t get a good fit of the height?


At the same time, I also used another data for inverse fitting, and the difference between it and the first one is that the elution gradient is steeper.
Different from the above process, the experimental data can coincide well with the fitting data after the search scope is narrowed, but there is still a large sample peak. How can I improve this? As different experiments of the same sample, why are the NU and SIGMA values of the two experiments so different?