As far as I know particle porosity has an inverse proportion to the adsorption capacity of a particle due to how CADET is coded.
So an increase in particle porosity, which should mean a higher capacity due to more surface area inside the particle, would lead to a decrease in capacity in CADET simulations.
The question for me is how do I correct this in my model setup.
At the moment I am using a Gaussian distribution as basis for my porosity distribution.
With a homogeneous particle distribution inside the column this is not a issue, but I am also adding using position distributions. This means that my adsorption capacity is a function that is based on the particle capacities at column height x.
In the following test case I look at 3 columns:
c_feed = [10]
n_bound = [1]
t_in_seconds =300000*60
n_col= 4
volume_flow_rate = 4.71e-5
resolution = int(t_in_seconds/60)
adsorption_parameters = Dict()
adsorption_parameters.is_kinetic = 1
adsorption_parameters.FLDF_KKIN = [1]
adsorption_parameters.FLDF_KF = [41312.942982]
adsorption_parameters.FLDF_N = [2.092987]
k_film = [1]
porediffusion= [1]
k_RL = [0]
Reference column (blue):
sizeclasses= 1.25/2000
porosityclasses= 0.455
n_partype=1
vol_frac =1
Green graph (low porosity at the top)
sizeclasses= [1.25/2000,1.25/2000]
porosityclasses= [0.355,0.555]
n_partype=2
vol_frac =[1,0,1,0,0,1,0,1]
Red graph (high porosity at the top)
sizeclasses= [1.25/2000,1.25/2000]
porosityclasses= [0.355,0.555]
n_partype=2
vol_frac =[0,1,0,1,1,0,1,0]
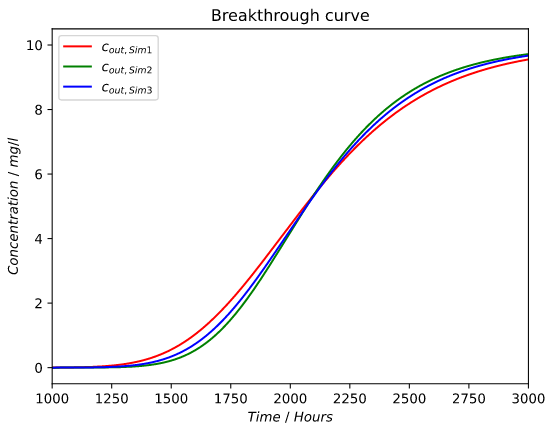
As mentioned before the graph (green) with a low porosity at the beginning/top of the column has a later initial breakthrough which would mean a higher adsorption capacity in the top part of the column.
One can also see that this effect does not seem symmetric.
Now for my case with a Gaussian distribution my Idea would be to mirror the volume fractions inside my column because the porosity distribution is symmetrical at the mean value.
Since my testcase I am not sure anymore if this approach is a valid correction.
So my question would be how I can adjust this issue numerically in my model setup for symmetrical and non-symmetrical porosity distributions.