I’m fitting an SMA + LumpedRateModelWithPores on an AKTA-generated LGE data with low loadings with CADET-Process. I’m currently not using reference concentrations as I want to see if I can re-use the previously fitted parameters from GoSilico/ChromX, which produced a very good simulation. I have verified that the Yamamoto’s results from both GoSilico & CADET-Process are in agreement. Other parameters (axial dispersion, film diffusion, etc…) are copy-pasted with proper unit conversions from GoSilico.
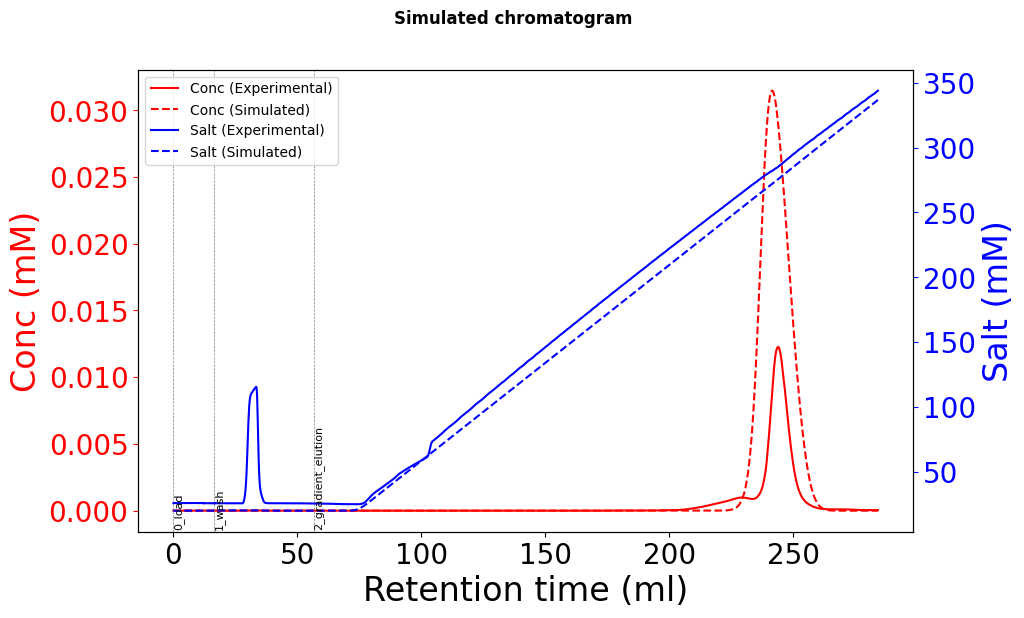
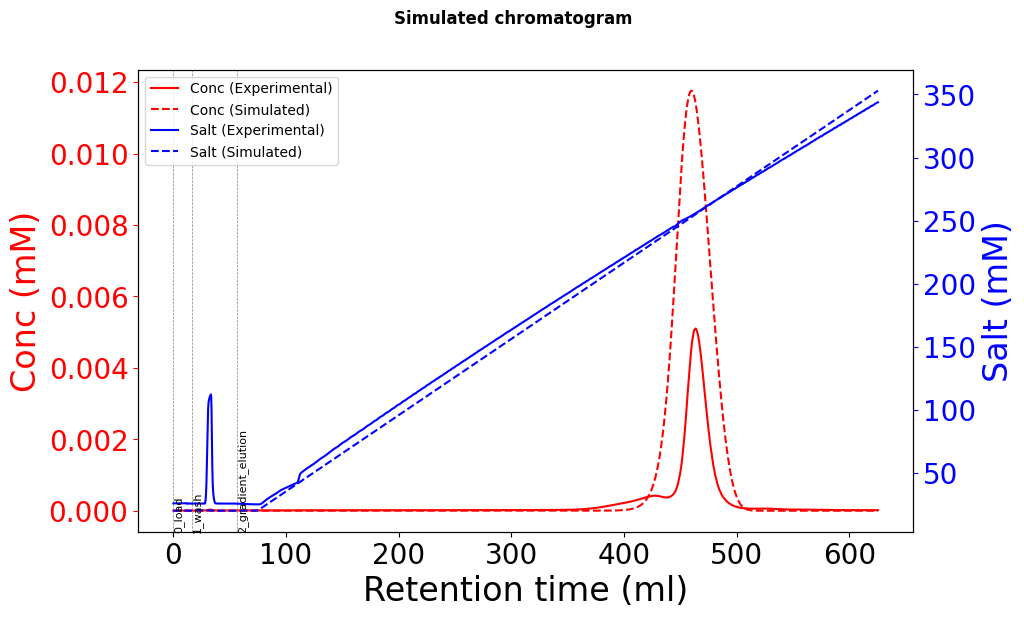
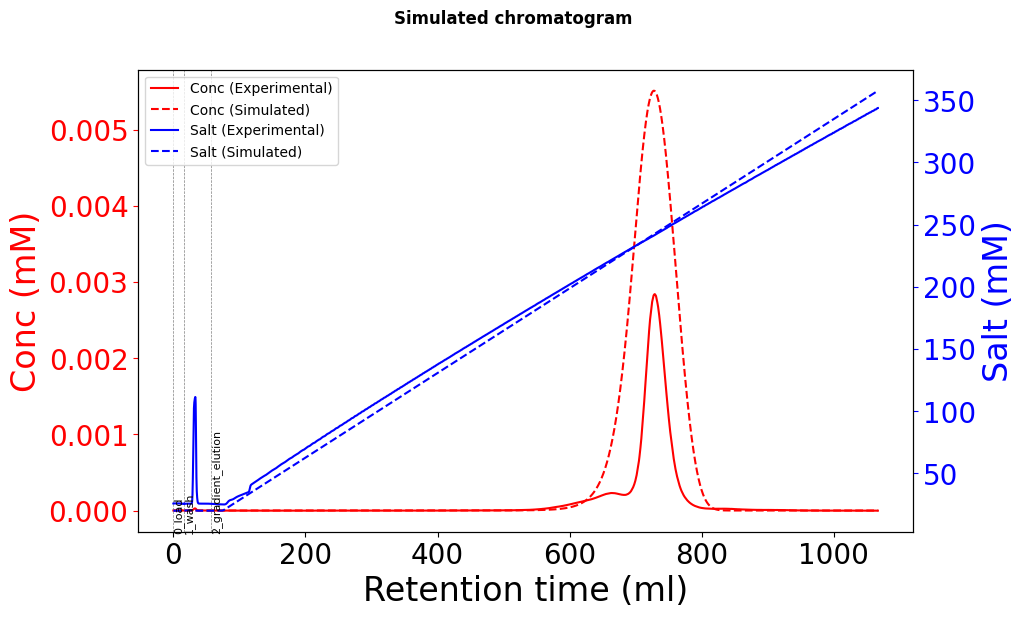
Using these Yamamoto-fitted parameters (k_eq, charge, steric factor), the elution peaks are in the right time position, but the height is approximately 2-3 times the actual data, and there seems to be peak broadening.
Just from your general curve-fitting experience, which other parameters could be re-fitted to adjust the simulated curve heights? Or should I just re-fit the whole model from scratch with inverse-fitting / CADET-Match?
We have already successfully converted ChromX parameters to CADET once (generalized ion-exchange isotherm). So I can say that this generally works.
What does the fit with ChromX look like?
Do you correctly convert the sample inlet profile (CADET uses mM by default)?
Due to conservation of mass, the injected mass must be the same as the eluted mass (assuming that everything comes off the column). The simulated chromatogram has a much greater mass than the experimental one.
Like @s.leweke said, it is critical that we check the mass balance (mass loaded must equal mass eluted for 100% recovery). A common issue when fitting experimental data is not accounting for UV detector nonlinearity and saturation. You can check the mass balance for your experimental data, and if you find that there is less in elution then that is your issue.
The easiest way around this is to use a higher wavelength, such at 295 nm instead of 280 nm. You can record three at once on the AKTA - I have used 280, 295, and 302. You just need to determine the extinction coefficient at different wavelengths by simply using UV ratios.
You can also purchase a 0.5 mm UV flow cell. Generally, I have found that above 1500 mAU the absorbance response to concentration becomes nonlinear. So either you can stay below this threshold or generate a UV calibration curve.
Calibration can be done by injecting protein samples of known concentration and correlating the resulting UV value with the concentration. You can fit this to a second degree poly in excel or some other tool/language. Then you can use this for signal in the nonlinear detection range. But you should always stay below detector saturation.
If you need more details, I have a section (3.6) in my publication (attached) on it.
EDIT:
I noticed that the concentration units on your experimental data indicate that the signal should be linear for a typical 2 mm flow cell (0.005 mM = 1.125 g/L for an antibody with eps = 1.5). So the signal might be in the linear range but the conversion from AKTA UV to concentration may not be correct.